
How much does a clinical trial cost?
The average cost of phase 1, 2, and 3 clinical trials across therapeutic areas is around $4, 13, and 20 million respectively. Pivotal (phase 3) studies for new drugs approved by the Food and Drug Administration (FDA) of the United States cost a median of $41,117 per patient.
The above-mentioned cost estimates across therapeutic areas were reported by Aylin Sertkaya et al., in a report submitted to the U.S. Department of Health and Human Services, containing Table 1 below.
Table 1: Total Per-Study Costs (in $ Millions), by Phase and Therapeutic Area [a] [b]
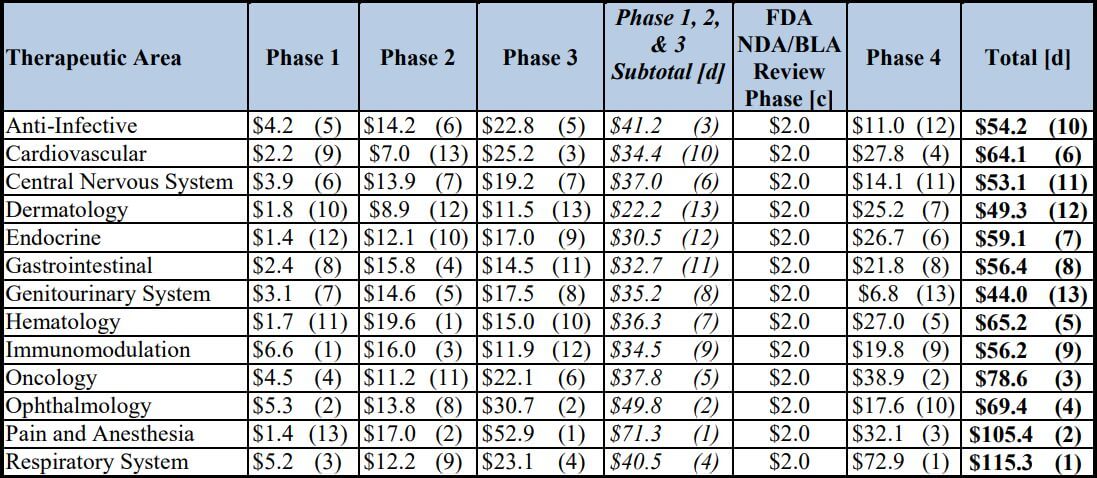
[a] The numbers in parentheses represent the rank in descending order.
[b] The cost for each phase assumes that a single trial (i.e., study) is conducted.
[c] The category represents the New Drug Application (NDA)/Biologic License Application (BLA) filing fee for an
application requiring clinical data and does not include any establishment or product fees that the filing entity might
need to pay in addition.
[d] Totals may not add up due to rounding
Per-patient pivotal trial costs were calculated by Thomas J. Moore et al., in their interesting study of 59 new therapeutic agents approved by the FDA from 2015 to 2016.
Factors impacting clinical trial costs
The cost of a clinical trial depends on several factors, such as study size (number of patients), locations (number of countries), number of clinical sites, therapeutic area, drug type, and the specific tests and procedures needed per protocol, among other aspects.
The study size (number of patients to be recruited) is closely related to the study phase. Early-phase (phase 1) trials require few patients (15-20 in average) while international phase 3 studies may involve hundreds of subjects (even thousands).
Furthermore, academic (non-commercial) and industry-led (commercial) trials should be distinguished. Academic, not-for-profit sponsors may conduct trials without commercial interest, counting on less financial resources to cover trial activities. On the other hand, commercial sponsors have higher budgets to execute their drug development programs.
Therefore, the clinical trial cost question does not have a single price answer. It really depends on the type, size, territory, and complexity of the research.
This article describes the general cost sections typically included in a Clinical Research Organization (CRO) quotation, including the total price of each section.
A typical CRO clinical trial budget: Understanding the main cost sections
CROs may structure their budgets in different ways, proposing different rates.
Nevertheless, clinical trial quotations normally include the following cost sections:
- Regulatory affairs
- Site identification and selection
- Site contracting and payments
- Site initiation and activation
- Site management
- Onsite monitoring
- Drug safety management
- Drug logistics
- Biological sample logistics
- Clinical supplies logistics
- Medical writing
- Site close-out
- Project management
- Study files/document management
- Data management
- Statistics
- Quality control
- Communication with central CRO/sponsor
- Pass-through costs
In the following paragraphs these cost items are further explained and valued.
Study example assumptions: A phase 3 trial in oncology
This article describes a budget example for a randomized, two-arm, phase 3 commercial clinical trial of two drugs (D1+D2 vs. D2 alone) in second line of advanced tumors.
A total of 350 patients would be recruited in 25 European sites, located in two countries (Spain and a second EU member state not specified).
Assumed trial stages and timelines are:
- Start-up: 6 months
- Recruitment: 36 months
- Per-patient treatment: 6 months
- Survival follow-up: 12 months for last patient in
- Close-out: 6 months
- Total study duration: 66 months
The proposed CRO management strategy consists of central management services provided for all countries, from the CRO headquarters located in Spain, and the use of senior Clinical Research Associates (CRAs) operating in the second European country for specific local tasks (mainly start-up, regulatory, and monitoring).
Regulatory affairs
Clinical trial regulatory affairs include the issuance of insurance policies in each country where the trial is conducted, ethicscommittee (EC) and regulatory authority (RA) initial submissions, study amendments, EC/RA reporting/communication, as well as the development and distribution of annual progress reports.
Total: $64,350
Site identification and selection
This section comprises the preparation, collection and analysis of feasibility questionnaires, as well as onsite selection visits, in order to verify the capabilities of each hospital.
Each selection visit includes time dedicated to scheduling, preparation, travel, the visit itself, post-visit report, and follow-up tasks.
Total: $46,200
Site contracting and payments
These tasks involve contract negotiation and execution with each hospital and site payment management (amounts paid by the Sponsor to the hospitals for each patient enrolled).
Total: $64,350 (the amounts paid per patient are listed as pass-through costs)
Site initiation and activation
The activation of clinical sites requires onsite initiation visits in which the CRAs and Clinical Project Managers of the CRO explain the goals and procedures of the trial to the site’s research team (Principal Investigator, Study Coordinator, nurses, etc.).
Each visit involves time dedicated to scheduling, preparation, travel, the visit itself, the post-visit report, and follow-up.
After the initiation visit, the activation process involves local document verification/signatures and providing sites with access to the different trial systems (EDC, etc.).
Total: $42,700
Site management
The CRAs of the CRO provide comprehensive support to sites during the recruitment and follow-up stages of the study.
This support consists of daily communication with sites via e-mail and telephone, responding to site inquiries, reviewing site performance, and escalating any issues to the Clinical Project Manager or Sponsor.
CRAs must also notify sites about any updated documents during the course of the study, ensuring that research teams have the latest versions of the study documents (e.g. Protocol, Patient Information Sheet / Informed Consent Form).
Total: $950,400
Onsite monitoring
Monitoring activities firstly require the development of a monitoring plan.
Then, onsite monitoring visits are conducted by CRAs during recruitment and follow-up, in order to verify source clinical data and other aspects, according to plan.
Each onsite monitoring visit includes time devoted to scheduling, preparation, travel, the visit itself, post-visit report, and follow-up.A reasonable monitoring strategy can be one onsite monitoring visit per site every 2 months.
Total: $1,069,670
Drug safety management
From the CRO’s side, drug safety requires the receipt, review, listing, reporting, and follow-up of serious adverse events (SAEs).
In addition, suspected unexpected serious adverse reactions (SUSARs) must be reported to regulatory authorities and other parties according to legislation.
The CRO can also take care of writing and distributing annual safety reports.
Another relevant role in drug safety is that of the Medical Monitor.
The CRO can provide a Medical Monitor for the trial, but this is not always the case since such role may also be played by staff of the pharma or biotech company.
Total: $29,820 (not including Medical Monitor)
Drug logistics
The tasks related to drug logistics may greatly vary depending on each trial.
For instance, the CRO may or may not be involved in helping with drug manufacturing and importation aspects.
The selection and contracting of a drug depot in charge of receiving, labelling, storing, and distributing the drugs to the sites is a commonly required task (subcontracted vendor).
The drug depot service cost is listed as a pass-through expense.
The CRO can also supervise drug stock availability (at depot and sites) and coordinate drug shipments from depot to sites.
Total: $55,790 (CRO cost not including depot subcontracted service)
Biological sample logistics
Clinical trials commonly require the handling of biological samples.
For example, oncology trials involve the collection and shipment of tumor and blood samples for translational/biomarker or pharmacokinetics (PK) studies.
CROs coordinate the shipment of biological samples from sites to central laboratories.
Total: $74,620 (not including shipping costs, which are listed as pass-through costs)
Clinical supplies logistics
Different trials may require different types of clinical supplies.
In cancer trials, laboratory kits are necessary for sites to collect and send biological samples (i.e. blood tubes, boxes).
CROs prepare and ship clinical supplies for sites as needed, tracking and sending additional supplies during the course of the study.
The supplies as such are listed apart, as pass-through costs.
Total: $22,575
Medical writing
Medical writing activities may include the writing and/or review of the study protocol, the Patient Information Sheet / Informed Consent Form, interim/final clinical study reports, and scientific publications (abstracts, posters, and manuscripts), among other documents.
Total: $24,960
Site close-out
The closure of the clinical trial requires onsite close-out visits (one per site), used to review study documentation and materials to be archived, once the trial has ended.
An onsite close-out visit includes time for scheduling, preparation, travel, visit, post-visit report, and follow-up.
Total: $40,950
Project management
Clinical Project Managers are senior staff who develop the global project plan (including timelines and milestones), the central filing, and the communication plan.
Project Managers lead teams and have periodic meetings with other CRO staff, the Sponsor, and vendors.
They manage and supervise the project, including financial, clinical, technical, and administrative aspects.
In addition, Clinical Project Managers supervise, review, and follow up site initiation, monitoring, and close-out visit reports.
Total: $1,021,120
Study files/document management
CROs have Clinical Trial Assistants (CTAs) in charge of study file/document management.
These tasks encompass Investigator Site File (ISF) preparation and shipments, electronic Trial Master File (eTMF) maintenance, and final eTMF reconciliation.
Total: $148,993
Data management
The CRO’s Data Managers develop the data management plan and take care of the electronic Case Report Form (eCRF) or Electronic Data Capture (EDC) system specification, configuration, development, testing, and validation.
Moreover, Data Managers provide EDC technical support to sites, perform data coding (adverse events, concomitant medications, and medical history), and review/clean EDC data (through data queries).
In addition, data management activities include SAE reconciliation with clinical database, as well as database locks and exports.
Total: $435,851
Statistics
Biostatisticians and statistical programmers will work on the development of the randomization code (if the trial is randomized) and the statistical analysis plan.
They will also work on the sample size calculation, protocol review, SAS programming, and statistical analyses and reports.
If required, statistical programmers will be in charge of SDTM/ADaM dataset specifications, mapping, programming, and validation.
Total: $150,518
Quality control
In order to ensure the quality of the study, the following activities will be conducted by the CRO’s Quality Managers: eTMF/document reviews, protocol deviation management, and hosting/attending Sponsor’s audits and regulatory inspections at the CRO/site.
Total: $70,460
Communication with central CRO/sponsor
When the CRO uses CRAs in other countries, these CRAs need to dedicate time to communicate with the CRO’s central office as well as with the Sponsor. This may include onsite meetings and teleconferences.
Total: $89,100
Pass-through costs
Pass-through costs are those expenses and services related to other parties or vendors.
In clinical trials, typical pass-through costs include:
- Trial insurance policies for each country
- Shipping: physical files to sites, site contracts, and tumor/blood samples
- Blood tubes and shipping packages
- Office supplies: files, paper, and printing
- Payments to sites per enrolled patient (to cover clinical procedures and laboratory tests)
- Publication fees
- Ethics committee evaluation fees
- Site contract fees
- Regulatory authority evaluation fees
- Travel costs for selection, initiation, routine monitoring, and close-out visits
- Central pathology and radiology reviews
- Translational/biomarker studies
- Coordinating investigators
- Drug manufacturing and testing
- Drug distribution services
- EDC license and service fees
- Web tools (imaging platforms, eTMF)
- Document translations
- Data and Safety Monitoring Board (DSMB)
Total: $8,526,728
CLINICAL TRIAL BUDGET TOTAL: $12,929,155
Conclusion
The phase 3 clinical trial budget example discussed in this article adds up to $12.9 million.
However, clinical trial costs are highly dependent on a long list of factors that can vary substantially.
For instance, a phase 3 clinical trial in oncology may recruit 350 patients while another phase 3, depending on the disease, may need to enroll more than 1,000 participants.
As general rule of thumb, the average cost of phase 1, 2, and 3 clinical trials across therapeutic areas is $4, 13, and 20 million respectively [1]. Pivotal studies cost a median of $41,117 per patient [2].
References:
[1] Examination of Clinical Trial Costs and Barriers for Drug Development:
https://aspe.hhs.gov/report/examination-clinical-trial-costs-and-barriers-drug-development
[2] Estimated Costs of Pivotal Trials for Novel Therapeutic Agents Approved by the US Food and Drug Administration, 2015-2016
https://jamanetwork.com/journals/jamainternalmedicine/fullarticle/2702287








